
Marketing Authorisation Holders (MAHs) are responsible for the quality, safety and efficacy of their products. Hence, a risk evaluation of all currently for EU market manufactured medicinal products containing chemically synthetized active pharmaceutical ingredients (APIs) are required to identify the risk of N-nitrosamine formation or (cross-)contamination. If as a result of the evaluation, a risk of presence of N-nitrosamines is identified, confirmatory testing should be carried out to confirm or refute the presence of N-nitrosamines.
The current regulation on N-nitrosamine impurities in medicinal products identifies three groups of risk factors:
- manufacturing process and storage of the API;
- manufacturing process and storage of the finished product;
- cross-contamination in shared facilities.
As it is known, N-nitrosamines can be formed when an amine and a nitrosating agent are combined under favourable conditions. Other pathways of generation of nitrosamines are oxidation and reduction processes from hydrazine-type compounds and N-nitroderivatives.
The initial and most important step in the risk evaluation of the presence of N-nitrosamines in the medicinal product is to identify if API and/or final product could be at risk of impurity formation, and what are the roots of possible contamination of the final product.
Currently identified risk factors for N-nitrosamine impurities in medicinal products are listed in specific EMA guidelines.
Table: N-nitrosamine formation routes and classification of reaction types (EMA 2020)
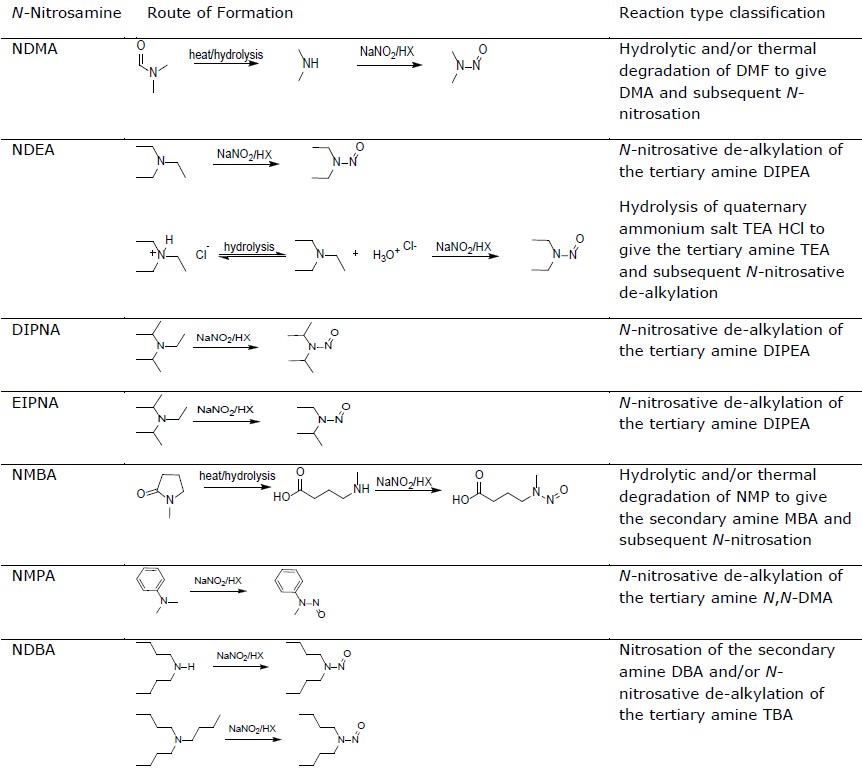
However, the list is not exhaustive and further root causes may also be applicable – it is up to MAH to determine if there is a risk with their product.
ACA can help you with the preparation of a risk assessment concerning the possible formation of N-nitrosamines.
Contact:
Bianca Leubner
Telephone: +49 (0) 341 223 292 36
email