
Der Marketing Authorisation Holder (MAH) ist für die Qualität, Sicherheit und Wirksamkeit seiner Produkte verantwortlich. Daher ist eine Risikobewertung aller derzeit für den EU-Markt hergestellten Arzneimittel, die chemisch synthetisierte pharmazeutische Wirkstoffe (API) enthalten, erforderlich, um das Risiko der Bildung von N-Nitrosaminen oder einer (Kreuz-)Kontamination zu ermitteln. Falls ein Risiko für das Vorhandensein von N-Nitrosaminen besteht, sollten Studien durchgeführt werden, um das Vorhandensein von N-Nitrosaminen zu bestätigen oder zu widerlegen.
In der geltenden Verordnung über N-Nitrosamin-Verunreinigungen in Arzneimitteln werden drei Gruppen von Risikofaktoren genannt:
- Herstellungsverfahren und Lagerung des APIs;
- Herstellungsverfahren und Lagerung des Fertigarzneimittels;
- Kreuzkontamination in Herstellungsstätten mit mehreren Produkten.
N-Nitrosamine können gebildet werden, wenn ein Amin und ein Nitrosierungsmittel unter günstigen Bedingungen zusammenkommen. Andere Wege zur Bildung von Nitrosaminen sind Oxidations- und Reduktionsprozesse aus hydrazinartigen Verbindungen und N-Nitroderivaten.
Der erste und wichtigste Schritt bei der Risikobewertung des Vorhandenseins von N-Nitrosaminen im Arzneimittel besteht darin, festzustellen, ob für den Wirkstoff und/oder das Fertigprodukt ein Risiko für die Bildung von Verunreinigungen bestehen könnte und welches die Ursachen für eine mögliche Verunreinigung des Fertigprodukts sind.
Die derzeit identifizierten Risikofaktoren für N-Nitrosamin-Verunreinigungen in Arzneimitteln sind in spezifischen EMA-Leitlinien aufgeführt.
Tabelle: N-Nitrosamin-Bildung und Klassifizierung der Reaktionstypen (EMA 2020)
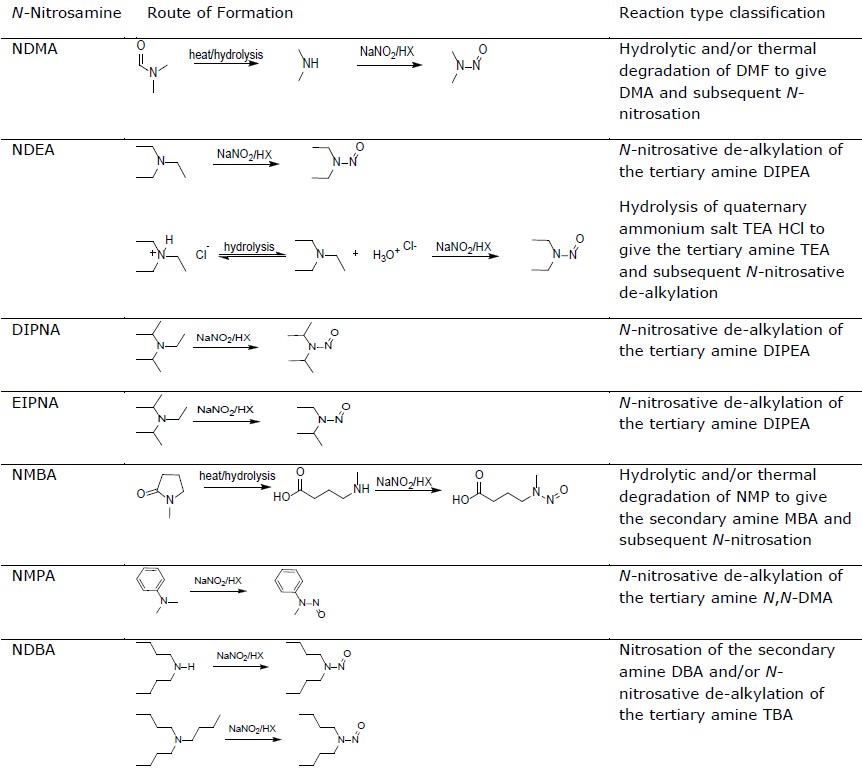
Die Liste ist jedoch nicht erschöpfend, und es können noch weitere Ursachen in Frage kommen – es obliegt dem MAH, festzustellen, ob bei seinem Produkt ein Risiko besteht.
Die ACA kann Ihnen bei der Erstellung einer Risikobewertung zur möglichen Bildung von N-Nitrosaminen helfen.
Kontakt:
Bianca Leubner
Tel.: +49 (0) 341 223 292 36
E-Mail